What is it?
Summary
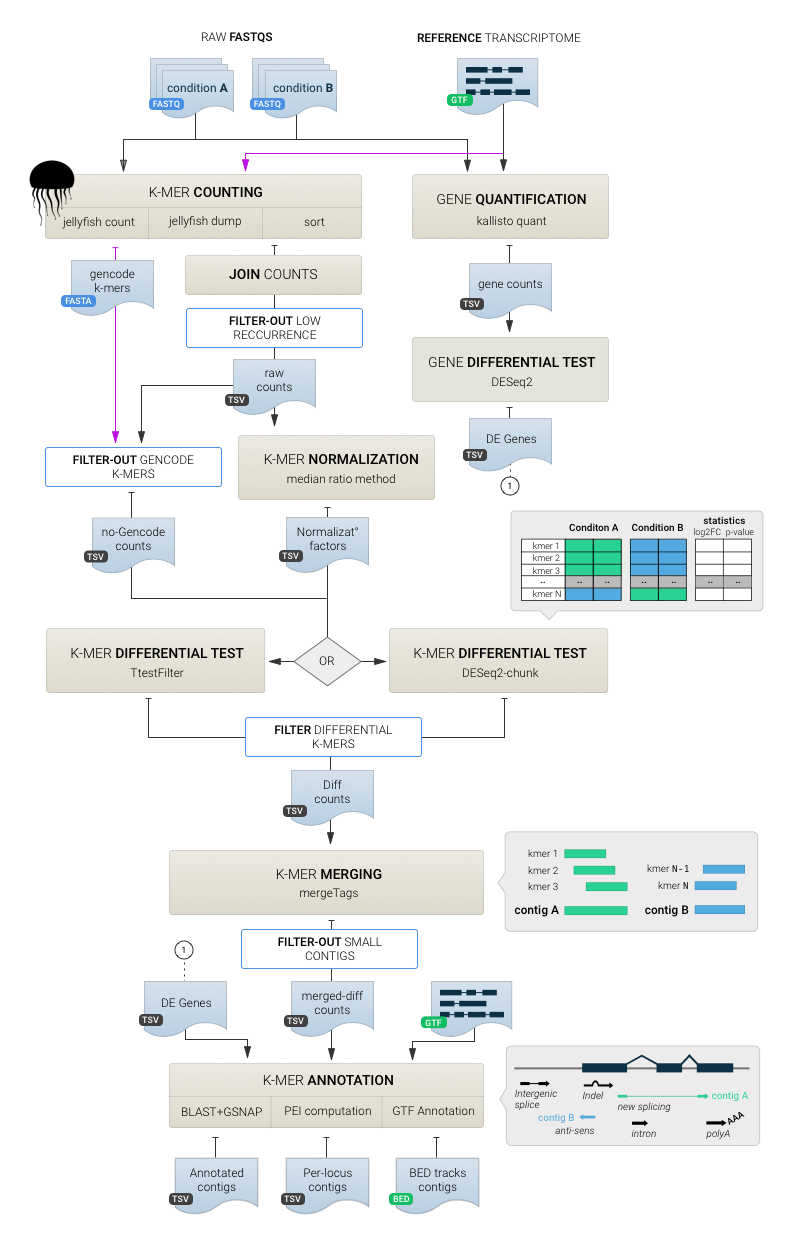
DE-kupl is a computational protocol that aims to capture all k-mer variation in an input set of RNA-seq libraries. This protocol is composed of four main components :
- Indexing: index and count all k-mers (k=31) in the input libraries
- Filtering: delete k-mers representing potential sequencing errors or perfectly matching known transcripts
- Differential Expression (DE): select k-mers with significantly different abundance across conditions
- Assembly and annotation: build contigs of assembled k-mers and annotate contigs based on sequence alignment.
Installation and usage
The DE-kupl project is composed of two sub-project:
- DE-kupl run which handle the DE-kupl procude from raw FASTQ to the assembly of differentially expressed k-mers.
- DE-kupl annotation which annotate DE contigs produced bu DE-kupl run.
Therfore, you need to download and execute both sub-projects.
WARNINGS: Currently DE-kupl, is set up for Human genome only. Manual modification of the sources is needed to handle other species.
DE-kupl pipeline
First, Jellyfish is applied to count k-mers in all libraries. K-mers counts are then joined into a count matrix and filtered for low-recurrence and matching to the reference transcriptome. Normalization factors are computed from raw K-mer counts and the DE procedure is applied. Finally overlapping DE k-mers are merged into contigs and annotated based on their alignment to reference and overlap with annotations. In parallel, FASTQs are processed with Kallisto to estimate gene-level counts and differentially expressed genes are derived using DESeq2. The list of DE genes is used for contig annotation only.